文章目录
FASTA和FASTQ文件,其实还是文本文件,用于存储序列信息。平时为了存储方便,节省空间,所以变成了GZ的压缩文件(二进制文件,不能直接用head 、less等命令直接查看)。之前没有好好记笔记,现在补上😑
FASTA是一种非常简单的生物序列储存格式,包括核酸序列(DNA/RNA)或者组成蛋白质的氨基酸序列(Amino Acid sequence,简称AA序列)。主要由交替重复的两部分(行)组成:
# 核酸序列
>gi|13650073|gb|AF349571.1| Homo sapiens hemoglobin alpha-1 globin chain (HBA1) mRNA, complete cds
CCCACAGACTCAGAGAGAACCCACCATGGTGCTGTCTCCTGACGACAAGACCAACGTCAAGGCCGCCTGG
GGTAAGGTCGGCGCGCACGCTGGCGAGTATGGTGCGGAGGCCCTGGAGAGGATGTTCCTGTCCTTCCCCA
CCACCAAGACCTACTTCCCGCACTTCGACCTGAGCCACGGCTCTGCCCAGGTTAAGGGCCACGGCAAGAA
GGTGGCCGACGCGCTGACCAACGCCGTGGCGCACGTGGACGACATGCCCAACGCGCTGTCCGCCCTGAGC
GACCTGCACGCGCACAAGCTTCGGGTGGACCCGGTCAACTTCAAGCTCCTAAGCCACTGCCTGCTGGTGA
CCCTGGCCGCCCACCTCCCCGCCGAGTTCACCCCTGCGGTGCACGCCTCCCTGGACAAGTTCCTGGCTTC
TGTGAGCACCGTGCTGACCTCCAAATACCGTTAAGCTGGAGCCTCGGTGGCCATGCTTCTTGCCCCTTTG
G
# 蛋白序列
>sp|P69905|HBA_HUMAN Hemoglobin subunit alpha OS=Homo sapiens GN=HBA1
MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHG
KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTP
AVHASLDKFLASVSTVLTSKYR
FASTQ文件其实就是FASTA文件的扩展,把测序的质量信息也存储在文件中。在这种格式中,序列和质量分数都表示为单个ASCII字符。主要由四个包含不同信息的交替重复部分(行)组成:
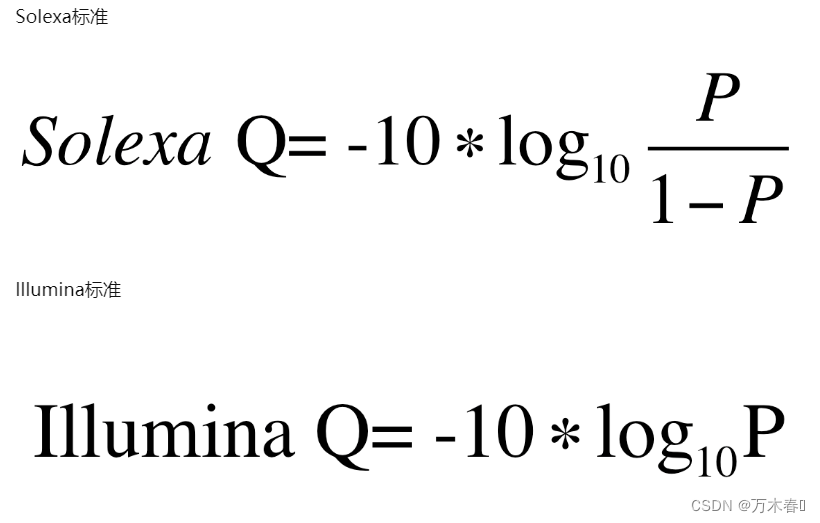
计算方式的细节,参见:https://zhuanlan.zhihu.com/p/20714540
见图:

了解了二者的格式,格式的转换就相对容易,方式的选择就会多种多样。
sed可以用来选择性的输出所需的行,所以,只要在Fastq文件的每4行中,选择打印第1、2行,就可以完成FASTQ到FASTA的转换:
sed -n '1~4s/^@/>/p;2~4p' INFILE.fastq > OUTFILE.fasta
如果不太好理解,也可以配合paste命令,将FASTQ的每四行排列为以制表符分隔的一行,然后使用sed + tr命令转换:
cat INFILE.fastq | paste - - - - |cut -f 1, 2| sed 's/@/>/'g | tr -s "/t" "/n" > OUTFILE.fasta
有了转换思路,尝试用更全面的awk命令将FASTQ转换为FASTA(与其说是命令,我更倾向awk是一种语言):
cat infile.fq | awk '{if(NR%4==1) {printf(">%s\n",substr($0,2));} else if(NR%4==2) print;}' > file.fa
简单解释一下,NR代表记录数,循环当中可以简单的理解为 “ 第几行 ” ,或者说是 “ 行号 ” 。在END块内使用,就是文件总记录数(差不多就是总行数)。那么,NR%4就是行号除以4的余数,head -n 8 tmp.fq | awk '{printf("Line:%s remainder: %s\ncontext: %s\n", NR, NR%4, $0)}':分别显示行号(Line),行号除以4的余数(remainder)和该行的内容(context)
Line:1 remainder: 1
context: @A00234:1023:HLGC5DSX3:1:1101:1253:1000 1:N:0:ATCGTACT
Line:2 remainder: 2
context: NGATTGATCGATGAGTAAACAAAGTGAGCAACAGACAGAAAAGGGGCTGTCTCTTATACACATCTCCGAGCCCACGAGACATCGTACTATCTCGTATGCCGTCTTCTGCTTGAAAAATGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG
Line:3 remainder: 3
context: +
Line:4 remainder: 0
context: #FFFFFFFFFFFFFFFFFFFFFFFFFFFF:FFFF,FFFFFFFFFFF,FFFFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFF:FFFFF,FFFFF,,F,FF:F,FF:F,,,,:,,,:::F,:FFFF:FFFFFFFFFFFFFFFFFFFFF
Line:5 remainder: 1
context: @A00234:1023:HLGC5DSX3:1:1101:1542:1000 1:N:0:ATCGTACT
Line:6 remainder: 2
context: NTTTGATTCCGTACTTTGCCTATTCGGTATTCCGAGAGGCTGGTCGGATTGGGTTACAGCCTTGTCTGTATAGGAGCTGTCTCTTATACACATCTCCGAGCCCACGAGACATCGTACTATCTCGTATGCCCTCTTCTGCTTGAAAAATGG
Line:7 remainder: 3
context: +
Line:8 remainder: 0
context: #FFFFFFFFF:FFFFFFFF,FFFFFF:FFFFFFFFFFFFFFFFFFF,FF:FFFFFFF:FFF:FFFFFFFFFFFFFFFF:FFFFFFFFFFFFF:FFFFFFFFFFFFFFFFFFFFFFFF::FFFFFFF,:,F,:FF,,,,,,F,F,FF,,,,
可以看到当余数为1或者2的时候,就是转换需要的,所以此时使用判断语句,当余数等于 1 或者 2 的时候,打印需要的内容。这里还使用了awk内置函数substr,语法:substr(input-string, location, length),为了去除@符号,所以就从序列的第二位开始打印。
Bioawk & Seqtk
来自李恒大佬开发的工具,使用方式:
bioawk -c fastx '{print ">"$name"\n"$seq}' input.fastq > output.fasta
seqtk seq -a input.fastq > output.fasta
可以使用压缩文件 (.gz) 作为输入
EMBOSS:seqret
使用方式:
seqret -sequence reads.fastq -outseq reads.fasta
FASTX-toolkit
fastq_to_fasta函数,适合超大数据:
fastq_to_fasta -h
usage: fastq_to_fasta [-h] [-r] [-n] [-v] [-z] [-i INFILE] [-o OUTFILE]
# Remember to use -Q33 for illumina reads!
version 0.0.6
[-h] = This helpful help screen.
[-r] = Rename sequence identifiers to numbers.
[-n] = keep sequences with unknown (N) nucleotides.
Default is to discard such sequences.
[-v] = Verbose - report number of sequences.
If [-o] is specified, report will be printed to STDOUT.
If [-o] is not specified (and output goes to STDOUT),
report will be printed to STDERR.
[-z] = Compress output with GZIP.
[-i INFILE] = FASTA/Q input file. default is STDIN.
[-o OUTFILE] = FASTA output file. default is STDOUT.
了解前面,也知道,实现这种转换的关键是质量信息,但是,咱没有,所有这时候就需要模拟一个,已经有一些工具帮咱实现了类似功能,比如python工具pyfastaq、bioconvert等,代码也很简单,比如:
# pyfastaq 默认质量40
fastaq to_fake_qual in.fasta - | fastaq fasta_to_fastq in.fasta - out.fastq
# bioconvert 使用虚拟质量 "I", 并且第一行信息不加其他的东西
bioconvert fasta2fastq in.fa tout.fq
重要的是了解格式,掌握之后,格式之间的转换就会得心应手,至于用什么工具,什么语言写的工具,看自己喜欢喽,当然,对于生物信息学,Linux系统、命令行工具暂时还是绕不过去的,必须要掌握。
至于其他语言,只要能达成目的!!
我的目标是转换表单输入,例如“100兆字节”或“1GB”,并将其转换为我可以存储在数据库中的文件大小(以千字节为单位)。目前,我有这个:defquota_convert@regex=/([0-9]+)(.*)s/@sizes=%w{kilobytemegabytegigabyte}m=self.quota.match(@regex)if@sizes.include?m[2]eval("self.quota=#{m[1]}.#{m[2]}")endend这有效,但前提是输入是倍数(“gigabytes”,而不是“gigabyte”)并且由于使用了eval看起来疯狂不安全。所以,功能正常,
我正在尝试测试是否存在表单。我是Rails新手。我的new.html.erb_spec.rb文件的内容是:require'spec_helper'describe"messages/new.html.erb"doit"shouldrendertheform"dorender'/messages/new.html.erb'reponse.shouldhave_form_putting_to(@message)with_submit_buttonendendView本身,new.html.erb,有代码:当我运行rspec时,它失败了:1)messages/new.html.erbshou
我在从html页面生成PDF时遇到问题。我正在使用PDFkit。在安装它的过程中,我注意到我需要wkhtmltopdf。所以我也安装了它。我做了PDFkit的文档所说的一切......现在我在尝试加载PDF时遇到了这个错误。这里是错误:commandfailed:"/usr/local/bin/wkhtmltopdf""--margin-right""0.75in""--page-size""Letter""--margin-top""0.75in""--margin-bottom""0.75in""--encoding""UTF-8""--margin-left""0.75in""-
我在我的项目目录中完成了compasscreate.和compassinitrails。几个问题:我已将我的.sass文件放在public/stylesheets中。这是放置它们的正确位置吗?当我运行compasswatch时,它不会自动编译这些.sass文件。我必须手动指定文件:compasswatchpublic/stylesheets/myfile.sass等。如何让它自动运行?文件ie.css、print.css和screen.css已放在stylesheets/compiled。如何在编译后不让它们重新出现的情况下删除它们?我自己编译的.sass文件编译成compiled/t
我想将html转换为纯文本。不过,我不想只删除标签,我想智能地保留尽可能多的格式。为插入换行符标签,检测段落并格式化它们等。输入非常简单,通常是格式良好的html(不是整个文档,只是一堆内容,通常没有anchor或图像)。我可以将几个正则表达式放在一起,让我达到80%,但我认为可能有一些现有的解决方案更智能。 最佳答案 首先,不要尝试为此使用正则表达式。很有可能你会想出一个脆弱/脆弱的解决方案,它会随着HTML的变化而崩溃,或者很难管理和维护。您可以使用Nokogiri快速解析HTML并提取文本:require'nokogiri'h
我需要读入一个包含数字列表的文件。此代码读取文件并将其放入二维数组中。现在我需要获取数组中所有数字的平均值,但我需要将数组的内容更改为int。有什么想法可以将to_i方法放在哪里吗?ClassTerraindefinitializefile_name@input=IO.readlines(file_name)#readinfile@size=@input[0].to_i@land=[@size]x=1whilex 最佳答案 只需将数组映射为整数:@land边注如果你想得到一条线的平均值,你可以这样做:values=@input[x]
这道题是thisquestion的逆题.给定一个散列,每个键都有一个数组,例如{[:a,:b,:c]=>1,[:a,:b,:d]=>2,[:a,:e]=>3,[:f]=>4,}将其转换为嵌套哈希的最佳方法是什么{:a=>{:b=>{:c=>1,:d=>2},:e=>3,},:f=>4,} 最佳答案 这是一个迭代的解决方案,递归的解决方案留给读者作为练习:defconvert(h={})ret={}h.eachdo|k,v|node=retk[0..-2].each{|x|node[x]||={};node=node[x]}node[
我有一个对象has_many应呈现为xml的子对象。这不是问题。我的问题是我创建了一个Hash包含此数据,就像解析器需要它一样。但是rails自动将整个文件包含在.........我需要摆脱type="array"和我该如何处理?我没有在文档中找到任何内容。 最佳答案 我遇到了同样的问题;这是我的XML:我在用这个:entries.to_xml将散列数据转换为XML,但这会将条目的数据包装到中所以我修改了:entries.to_xml(root:"Contacts")但这仍然将转换后的XML包装在“联系人”中,将我的XML代码修改为
为了将Cucumber用于命令行脚本,我按照提供的说明安装了arubagem。它在我的Gemfile中,我可以验证是否安装了正确的版本并且我已经包含了require'aruba/cucumber'在'features/env.rb'中为了确保它能正常工作,我写了以下场景:@announceScenario:Testingcucumber/arubaGivenablankslateThentheoutputfrom"ls-la"shouldcontain"drw"假设事情应该失败。它确实失败了,但失败的原因是错误的:@announceScenario:Testingcucumber/ar
我在我的项目中添加了一个系统来重置用户密码并通过电子邮件将密码发送给他,以防他忘记密码。昨天它运行良好(当我实现它时)。当我今天尝试启动服务器时,出现以下错误。=>BootingWEBrick=>Rails3.2.1applicationstartingindevelopmentonhttp://0.0.0.0:3000=>Callwith-dtodetach=>Ctrl-CtoshutdownserverExiting/Users/vinayshenoy/.rvm/gems/ruby-1.9.3-p0/gems/actionmailer-3.2.1/lib/action_mailer