部署好相关的软件和工具
BWA (Burrow-Wheeler Aligner) Version 0.7.17-r1188 解压、编译
Samtools Version: 1.16.1
解压 tar jxvf samtools-1.16.1.tar.gz
进入目录 cd samtools-1.16.1
配置 ./configure --prefix=~/biosoft/samtools-1.16.1
编译安装 make
make install
Picard 直接下载java包picard.jar
GATK gatk-4.3.0.0 下载后不用编译直接使用
上述软件均需添加到环境变量
bwa index Sus_scrofa.Sscrofa11.1.dna.toplevel.fa
java -jar /home/dengsx/biosoft/picard.jar CreateSequenceDictionary \
R=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa \
O=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa.dict
samtools faidx Sus_scrofa.Sscrofa11.1.dna.toplevel.fa ##fai索引的构建
后续GATK的使用需要用到多种索引类型,要用多个软件创建
dict索引文件也可以通过gatk来获取
gatk CreateSequenceDictionary -R E.coli_K12_MG1655.fa -O E.coli_K12_MG1655.dict && echo "** dict done **"(本人没试过)
###需要注意的是.dict文件的名字前缀需要和fasta的一样,并跟它在同一个路径下,这样GATK才能够找到。
java -jar /home/dengsx/biosoft/gatk-4.3.0.0/gatk-package-4.3.0.0-local.jar IndexFeatureFile --input /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf > /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf.index.log 2>&1
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-hErFUiEQ-1666255841603)(Snipaste_2022-10-19_21-52-02.png)]
[外链图片转存失败,源站可能有防盗链机制,建议将图片保存下来直接上传(img-nuH0BtQJ-1666255841604)(Snipaste_2022-10-19_21-53-48.png)]
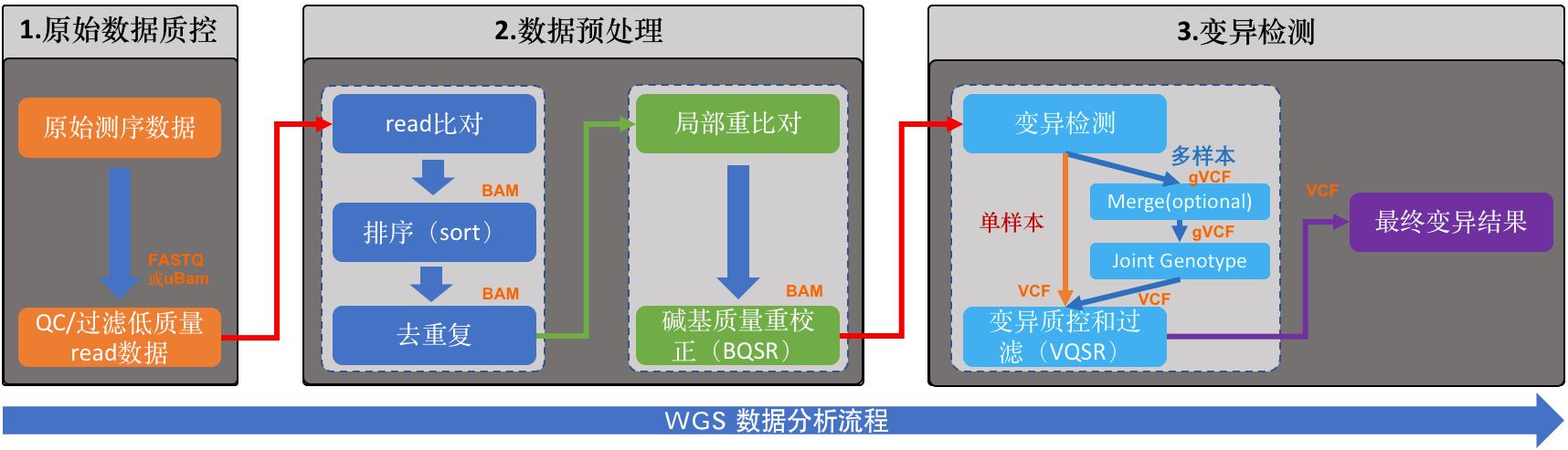
这一步完成之后,我们就可以将read比对至参考基因组了
将比对的输出结果直接重定向到一份*.sam文件中,这类文件是BWA比对的标准输出文件,。但SAM文件是文本文件,一般整个文件都非常巨大,因此,为了有效节省磁盘空间,用samtools将它转化为BAM文件(SAM的特殊二进制格式),而且BAM会更加方便于后续的分析。
bwa mem -t 5 -R '@RG\tID:YF62_E7\tPL:UNKNOWN\tLB:library2\tSM:YF62_E7' /home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa YF62_E7_1_clean.fq.gz YF62_E7_2_clean.fq.gz | samtools view -S -b - > YF62_E7.bam
-t,线程数,我们在这里使用4个线程;-R 接的是Read Group的字符串信息,这是一个非常重要的信息,以@RG开头,它是用来将比对的read进行分组的。不同的组之间测序过程被认为是相互独立的,这个信息对于我们后续对比对数据进行错误率分析和Mark duplicate时非常重要。
在Read Group中,有如下几个信息非常重要:
最好是在这一步就merge,后面的操作都是针对merge后的文件
samtools merge -o srr.bam srr1.bam srr2.bam
samtools sort -@ 1 -m 64G -O bam -o A65.sorted.bam A65.bam
其中,-@,用于设定排序时的线程数;-m,限制排序时最大的内存消耗,-O 指定输出为bam格式;-o 是输出文件的名字。建议在做类似分析的时候在文件名字将所做的关键操作包含进去,因为这样即使过了很长时间,当你再去看这个文件的时候也能够立刻知道当时对它做了什么。
for i in `ls *.sorted.bam`
do
java -jar /home/dengsx/biosoft/picard.jar MarkDuplicates \
REMOVE_DUPLICATES=false \
I=${input}/${i} \
O=${output}/${i%\.bam}.markup.bam \
M=${output}/${i%\.bam}.markup_metrics.txt
REMOVE_DUPLICATES=true建议使用第一种做法,只是标记出来,并留存这些序列,以便在你需要的时候还可以对其做分析。cd /home/dengsx/publicdata/markup_out
for markup_bam in `ls *.sorted.markup.bam`
do
samtools index ${markup_bam}
done
在重新校正碱基质量值(BQSR)之前把相同个体的bam文件merge,将同个样本的所有比对结果合并成唯一一个大的BAM文件
samtools merge -o srr.sorted.markup.bam srr1.sorted.markup.bam srr2.sorted.markup.bam
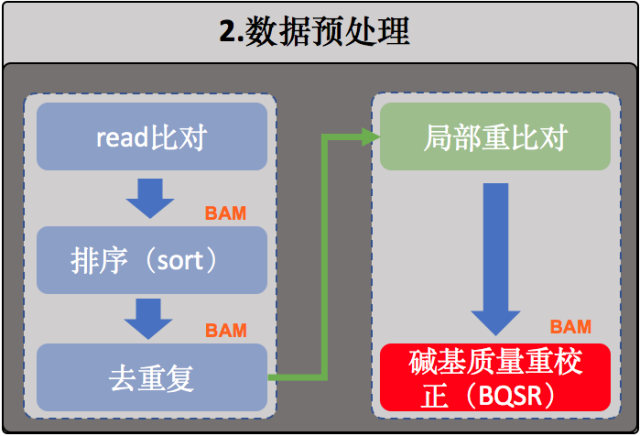
主要是通过机器学习的方法构建测序碱基的错误率模型,然后对这些碱基的质量值进行相应的调整。
这里包含了两个步骤:
第一步,BaseRecalibrator,这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
第二步,ApplyBQSR ,这一步利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
数据准备
dbSNP
数据处理
sus_scrofa.vcf.gz文件awk -F "\t" '!($8 ~ /\s/)' /home/dengsx/publicdata/dbsnp/sus_scrofa.vcf > /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf
java -jar /home/dengsx/biosoft/gatk-4.3.0.0/gatk-package-4.3.0.0-local.jar IndexFeatureFile --input /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf > /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf.index.log 2>&1
BQRS重新校正碱基质量值
fasta=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa
input=/home/dengsx/publicdata/markup_out/
gatk --java-options "-Xmx10G -Djava.io.tmpdir=./" BaseRecalibrator \
-R ${fasta} \
-I ${input}/A65.sorted.markup.bam \
--known-sites /home/dengsx/publicdata/dbsnp/sus_scrofa.nospace.vcf \
-O A65.recal_data.table
- 这里计算出了所有需要进行重校正的read和特征值,然后把这些信息输出为一份校准表文件(sample_name.recal_data.table)
# 不能只用bwa构建的索引来做BQSR
# 需要的索引类型很多(参考上面索引的构建)
.fai、.dictjava -jar /home/dengsx/biosoft/picard.jar CreateSequenceDictionary R=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa O=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa.dict
samtools faidx Sus_scrofa.Sscrofa11.1.dna.toplevel.fa
.idxfasta=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa
input=/home/dengsx/publicdata/markup_out/
bqsr=/home/dengsx/publicdata/BQSR/
gatk --java-options "-Xmx10G -Djava.io.tmpdir=./" ApplyBQSR \
-R ${fasta} \
-I ${input}/A65.sorted.markup.bam \
--bqsr-recal-file ${bqsr}/A65.recal_data.table \
-O A65.sorted.markdup.BQSR.bam
- 这一步利用第一步得到的校准表文件(sample_name.recal_data.table)重新调整原来BAM文件中的碱基质量值,并使用这个新的质量值重新输出一份新的BAM文件。
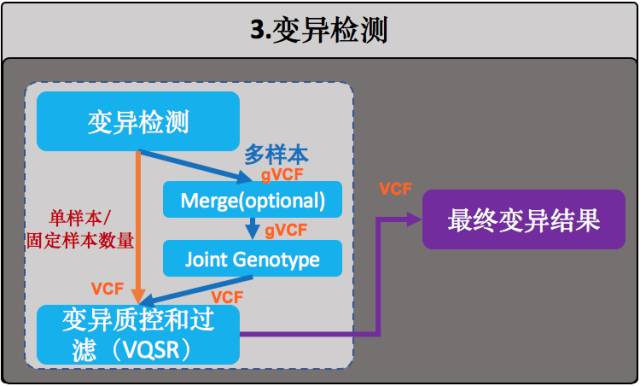
我们这里使用GATK HaplotypeCaller模块对样本中的变异进行检测,它也是目前最适合用于对二倍体基因组进行变异(SNP+Indel)检测的算法。
一般来说,在实际的WGS流程中对HaplotypeCaller的应用有两种做法,差别只在于要不要在中间生成一个gVCF:
基因组上各个不同的染色体之间其实是可以理解为相互独立的(结构性变异除外),也就是说,为了提高效率我们可以按照染色体一条条来独立执行这个步骤,最后再把结果合并起来就好了,这样的话就能够节省很多的时间
input=/home/dengsx/publicdata/BQSR
output=/home/dengsx/publicdata/haplotypeCaller
reference=/home/dengsx/publicdata/reference_genome/Sus_scrofa.Sscrofa11.1.dna.toplevel.fa
cd /home/dengsx/publicdata/haplotypeCaller
ls ../BQSR/*.sorted.markdup.BQSR.bam | while read line
do
gatk --java-options "-Xmx10G -Djava.io.tmpdir=./" HaplotypeCaller \
-I $input/${line##../BQSR/} \
-R $reference \
-ERC GVCF \
-ploidy 2 \
-O $output/${line##../BQSR/}.g.vcf > /home/dengsx/publicdata/haplotypeCaller/haplotypeCaller.log 2>&1
done
wait # this is necessary because both processes need to complete for the outside call to check on logs
echo $(date) done.main.process
参考:碱基矿工
我主要使用Ruby来执行此操作,但到目前为止我的攻击计划如下:使用gemsrdf、rdf-rdfa和rdf-microdata或mida来解析给定任何URI的数据。我认为最好映射到像schema.org这样的统一模式,例如使用这个yaml文件,它试图描述数据词汇表和opengraph到schema.org之间的转换:#SchemaXtoschema.orgconversion#data-vocabularyDV:name:namestreet-address:streetAddressregion:addressRegionlocality:addressLocalityphoto:i
有时我需要处理键/值数据。我不喜欢使用数组,因为它们在大小上没有限制(很容易不小心添加超过2个项目,而且您最终需要稍后验证大小)。此外,0和1的索引变成了魔数(MagicNumber),并且在传达含义方面做得很差(“当我说0时,我的意思是head...”)。散列也不合适,因为可能会不小心添加额外的条目。我写了下面的类来解决这个问题:classPairattr_accessor:head,:taildefinitialize(h,t)@head,@tail=h,tendend它工作得很好并且解决了问题,但我很想知道:Ruby标准库是否已经带有这样一个类? 最佳
我正在尝试使用Curbgem执行以下POST以解析云curl-XPOST\-H"X-Parse-Application-Id:PARSE_APP_ID"\-H"X-Parse-REST-API-Key:PARSE_API_KEY"\-H"Content-Type:image/jpeg"\--data-binary'@myPicture.jpg'\https://api.parse.com/1/files/pic.jpg用这个:curl=Curl::Easy.new("https://api.parse.com/1/files/lion.jpg")curl.multipart_form_
无论您是想搭建桌面端、WEB端或者移动端APP应用,HOOPSPlatform组件都可以为您提供弹性的3D集成架构,同时,由工业领域3D技术专家组成的HOOPS技术团队也能为您提供技术支持服务。如果您的客户期望有一种在多个平台(桌面/WEB/APP,而且某些客户端是“瘦”客户端)快速、方便地将数据接入到3D应用系统的解决方案,并且当访问数据时,在各个平台上的性能和用户体验保持一致,HOOPSPlatform将帮助您完成。利用HOOPSPlatform,您可以开发在任何环境下的3D基础应用架构。HOOPSPlatform可以帮您打造3D创新型产品,HOOPSSDK包含的技术有:快速且准确的CAD
本教程将在Unity3D中混合Optitrack与数据手套的数据流,在人体运动的基础上,添加双手手指部分的运动。双手手背的角度仍由Optitrack提供,数据手套提供双手手指的角度。 01 客户端软件分别安装MotiveBody与MotionVenus并校准人体与数据手套。MotiveBodyMotionVenus数据手套使用、校准流程参照:https://gitee.com/foheart_1/foheart-h1-data-summary.git02 数据转发打开MotiveBody软件的Streaming,开始向Unity3D广播数据;MotionVenus中设置->选项选择Unit
文章目录一、概述简介原理模块二、配置Mysql使用版本环境要求1.操作系统2.mysql要求三、配置canal-server离线下载在线下载上传解压修改配置单机配置集群配置分库分表配置1.修改全局配置2.实例配置垂直分库水平分库3.修改group-instance.xml4.启动监听四、配置canal-adapter1修改启动配置2配置映射文件3启动ES数据同步查询所有订阅同步数据同步开关启动4.验证五、配置canal-admin一、概述简介canal是Alibaba旗下的一款开源项目,Java开发。基于数据库增量日志解析,提供增量数据订阅&消费。Git地址:https://github.co
我正在尝试在Rails上安装ruby,到目前为止一切都已安装,但是当我尝试使用rakedb:create创建数据库时,我收到一个奇怪的错误:dyld:lazysymbolbindingfailed:Symbolnotfound:_mysql_get_client_infoReferencedfrom:/Library/Ruby/Gems/1.8/gems/mysql2-0.3.11/lib/mysql2/mysql2.bundleExpectedin:flatnamespacedyld:Symbolnotfound:_mysql_get_client_infoReferencedf
文章目录1.开发板选择*用到的资源2.串口通信(个人理解)3.代码分析(注释比较详细)1.主函数2.串口1配置3.串口2配置以及中断函数4.注意问题5.源码链接1.开发板选择我用的是STM32F103RCT6的板子,不过代码大概在F103系列的板子上都可以运行,我试过在野火103的霸道板上也可以,主要看一下串口对应的引脚一不一样就行了,不一样的就更改一下。*用到的资源keil5软件这里用到了两个串口资源,采集数据一个,串口通信一个,板子对应引脚如下:串口1,TX:PA9,RX:PA10串口2,TX:PA2,RX:PA32.串口通信(个人理解)我就从串口采集传感器数据这个过程说一下我自己的理解,
SPI接收数据左移一位问题目录SPI接收数据左移一位问题一、问题描述二、问题分析三、探究原理四、经验总结最近在工作在学习调试SPI的过程中遇到一个问题——接收数据整体向左移了一位(1bit)。SPI数据收发是数据交换,因此接收数据时从第二个字节开始才是有效数据,也就是数据整体向右移一个字节(1byte)。请教前辈之后也没有得到解决,通过在网上查阅前人经验终于解决问题,所以写一个避坑经验总结。实际背景:MCU与一款芯片使用spi通信,MCU作为主机,芯片作为从机。这款芯片采用的是它规定的六线SPI,多了两根线:RDY和INT,这样从机就可以主动请求主机给主机发送数据了。一、问题描述根据从机芯片手
前言一般来说,前端根据后台返回code码展示对应内容只需要在前台判断code值展示对应的内容即可,但要是匹配的code码比较多或者多个页面用到时,为了便于后期维护,后台就会使用字典表让前端匹配,下面我将在微信小程序中通过wxs的方法实现这个操作。为什么要使用wxs?{{method(a,b)}}可以看到,上述代码是一个调用方法传值的操作,在vue中很常见,多用于数据之间的转换,但由于微信小程序诸多限制的原因,你并不能优雅的这样操作,可能有人会说,为什么不用if判断实现呢?但是if判断的局限性在于如果存在数据量过大时,大量重复性操作和if判断会让你的代码显得异常冗余。wxswxs相当于是一个独立